Approaches used to solve the RNA Folding problem
Dynamic Programming using single parameter
The challenge arises when attempting to formulate a recurrence that defines OPT(j) based on solutions to smaller subproblems. Progress can be made to a certain extent: within the optimal secondary structure on b1b2 ... bj, one of the following holds true:
- j isn't engaged in a pair, or
- j forms a pair with t for a t < j - 4.
The Following inferences can be drawn from the above points :
- In the initial scenario, we simply refer to our solution for OPT(j - 1).
- In the subsequent scenario, due to the noncrossing constraint, it's evident that no pair can have one end positioned between 1 and t − 1 while the other end falls between t + 1 and j − 1. Consequently, we've effectively identified two distinct subproblems: one concerning the bases b1b2 ... bt−1 and the other concerning the bases bt+1 ... bj−1. While the former is addressed by OPT(t − 1), the latter isn't included in our list of subproblems since it doesn't commence with b1.
This realization prompts us to introduce an additional variable. We require the ability to handle subproblems that don't commence with b1; hence, we must consider subproblems on bibi+1 ... bj for all i ≤ j.
Dynamic Programming over intervals
Let OPT(i, j) denote the maximum number of base pairs in a secondary structure on bibi+1 ... bj. The no-sharp-turns condition lets us initialize OPT(i, j) = 0 whenever i ≥ j − 4.
Now, in the optimal sub-structure on bi,bi+1,...,bj, we have the same alternatives as before :
- j isn't engaged in a pair, or
- j forms a pair with t for a t < j - 4.
The Following inferences can be drawn from the above points :
- In the first case, we have OPT(i, j) = OPT(i, j - 1).
- In the second case, we recur on the two subproblems OPT(i, t - 1) and OPT(t + 1, j - 1), the noncrossing condition has isolated these two subproblems from each other.
Recurrence Relation
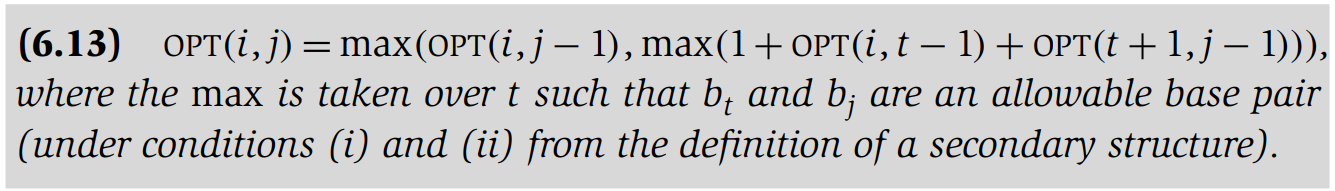
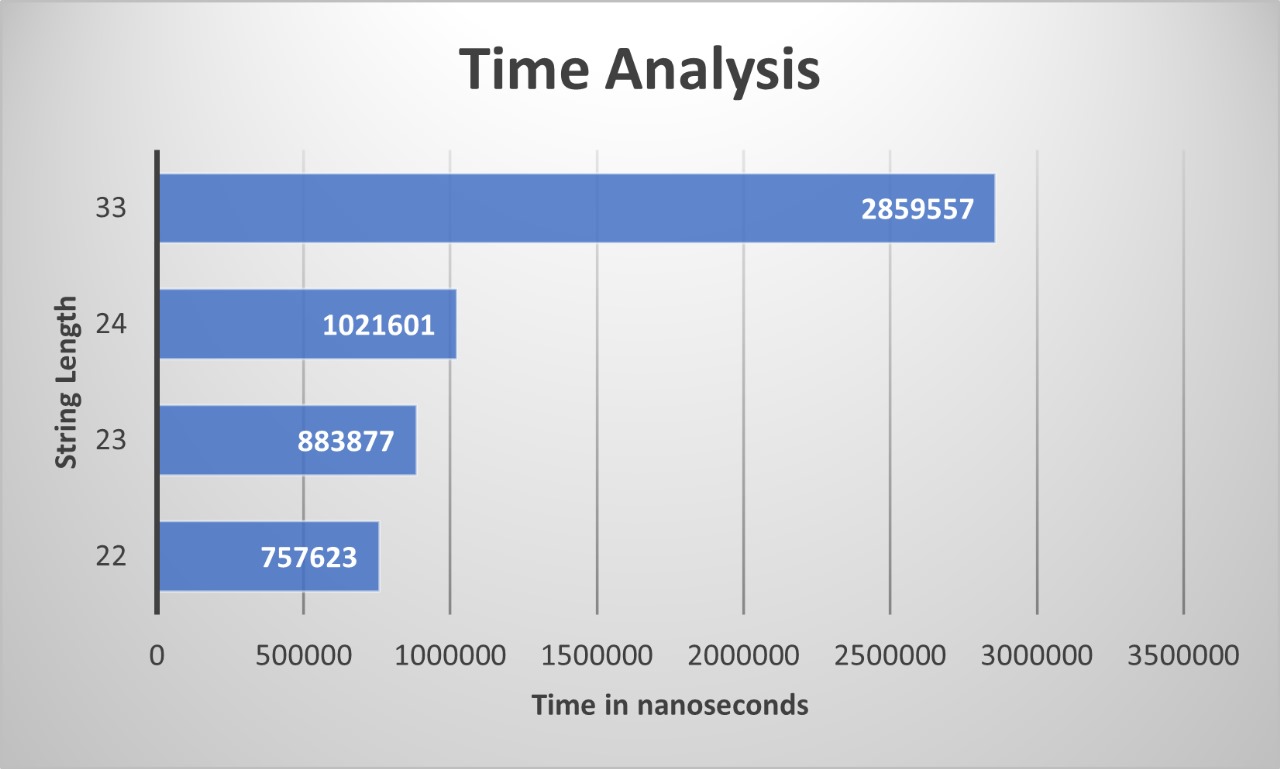
It is easy to bound the running time: there are O(n^2) subproblems to solve, and evaluating the recurrence takes time O(n) for each. Thus the running time is O(n^3).